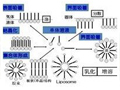
近年來,以聚乙二醇單甲醚( MPEG)和長鏈脂肪 醇為側鏈的梳型兩親聚合物,因其獨特性質及應用前景引起了理論研究領域和工業界的高度重視。此類聚合物大分子的結構類似梳子,在分散體系中由于其分子內部的微觀相分離而富集在表/界面,從而降低表/界面張力。在乳液和分散體系的穩定、表面改性和生物制藥等應用領域均具有明顯的優勢。其制備方法主要有兩種:大分子單體法和主鏈接枝法。大分子單體法是首先制備出具有反應性基團的大分子單體,然后將大分子單體和小分子化合物共聚得到梳型兩親聚合物。該法易于得到結構較規整的聚合物,分子結構的可設計性好,缺點是不易控制主鏈的結構和相對分子質量,而且大分子單體的分離純化過程較為繁瑣,產品的穩定性差。主鏈接枝法是先制備出適當結構的主鏈聚合物,然后和MPEG或長鏈脂肪醇通過酯化或酯交換反應制備出梳型兩親聚合物。該法容易調節主鏈、側鏈的結構和相對分子質量,缺點是所制備的接枝聚合物接枝率低,主鏈聚合反應和接枝反應需要分別進行,兩步反應可能引入不同溶劑造成污染。若調整主鏈接枝法的制備工藝,在兩步反應中采用相同的反應溶劑,那么通過“一鍋”反應便可以制備出目標產物,可以極大地節約成本,簡化生產工藝,有利于實現工藝的市場化和工業化。
作者采用主鏈接枝法,在兩步反應中使用相同的溶劑,嘗試用“一鍋”法將MPEG或/和十二醇接枝到苯乙烯.馬來酸酐共聚物( SMA)上,分別制備出了梳型兩親聚合物SMA-g-MPEG、SMA-g-C12H250H和SMA-g-MPEG+ C12H250H,并研究了它們的水溶性及其水溶液的表面活性。
1實驗部分
1.1試劑與儀器
苯乙烯( St),AR,北京市化學試劑公司,使用前用w( NaOH) =5%的水溶液洗滌兩次,無水Na2S04干燥24 h,減壓蒸餾后備用;馬來酸酐(MA),AR,天津市化學試劑公司,氯仿重結晶精制;偶氮二異丁腈( AIBN),CP,上海試四赫維有限公司,體積分數95%的乙醇重結晶精制,避光干燥保存;十二醇,AR,北京化工廠;MPEG,工業品,遼寧科隆化學品有限公司,平均相對分子質量1000;氫氧化鈉,AR,北京化工廠;四氫呋喃( THF),AR,天津科密歐化學試劑有限公司,經金屬鈉除水并常壓蒸餾;乙醚,AR,天津市化學試劑公司;對甲苯磺酸(PTSA),AR,北京化工廠。
K12表面張力儀,德國KrLiss公司;270 - 30型紅外光譜掃描儀,日本Hitachi公司;DRX - 300核磁共振儀,德國Bruker公司;Waters 1515凝膠滲透色譜儀,美國Waters公司。
1.2梳型兩親聚合物的制備
在裝有機械攪拌裝置、溫度計、N2保護裝置和冷凝回流裝置的四口燒瓶中,加入
梳型兩親聚合物的合成路線如下:

1.3梳型兩親聚合物的結構表征
主鏈聚合物SMA與接枝產物的結構用FTIR和1HNMR進行表征。
紅外光譜( FTIR):取少量樣品用丙酮溶解,KBr涂片,加熱除去丙酮。用日本Hitachi 270-30型傅立葉變換紅外光譜儀測定。
核磁共振氫譜(1HNMR):以氘代二甲基亞砜( DMSO)為溶劑,用美國Varian公司的DRX - 300Hz核磁共振儀測定,四甲基硅烷(TMS)作內標。
主鏈聚合物SMA與接枝產物的相對分子質量(簡稱分子量,下同)及其分布用凝膠滲透色譜儀進行表征。測量條件為:淋洗劑THF,流速1 mUmin,柱溫
1.4梳型兩親聚合物的性能測定
精確稱取一定量的聚合物樣品,放置到小燒杯中,加入適量去離子水并用NaOH溶液調節pH,于(25.0±0.1)℃的恒溫水浴中,在磁力攪拌下觀察其溶解性。若樣品未能溶解,則適當加熱并調節pH,再觀察其溶解性。
配制系列不同質量濃度的樣品水溶液,于(25.0±0.1)℃用Wilhelmy板法測定各溶液的表面張力r。以r對質量濃度(p)作圖,得到r-p表面張力曲線。
2結果與討論
2.1 梳型兩親聚合物的制備
實驗開始以THF為溶劑,通過St與MA的溶液共聚制備了黏均分子量在50000~70000的主鏈聚合物SMA。由于THF沸點較低(
2.2梳型兩親聚合物的結構表征
SMA和3種接枝聚合物的FTIR譜圖如圖1所示。比較SMA和接枝聚合物的紅外譜圖可發現,3種接枝聚合物的譜圖中均出現了1100~
對于SMA-g-MPEG,圖中63.35(d)為MPEG上端甲基中H的化學位移,63.54(b)為MPEG上乙氧基中H的化學位移,66.41~7.32(a)為St苯環中H的化學位移,62.51(e)為溶劑氘代DMSO中H的化學位移,由于氘代DMSO吸水性較強,譜圖里會出現水峰,63.43(c)便為其所含水中H的化學位移。1HNMR和FTIR的分析結果相結合,進一步證明SMA與MPEG通過酯化反應制備出了目標產物。在其IHNMR譜圖中,通過6=6.41~7.32的質子峰面積與6=3.54的質子峰面積的比值,可以得到MPEG在SMA-g-MPEG中的摩爾分數為23.8%。對于SMA-g-MPEG+C12 H250H,譜圖中63.35(d)、3.54(b)、2.51(e)、3.43(c)和6.41—7.32(a)處化學位移的歸屬與SMA-g-MPEG譜圖中相同,不同之處在于出現了60.90(g)處十二醇上端甲基中H的化學位移,61.22~1.37(f)處十二醇上亞甲基中H的化學位移。1HNMR和FTIR的分析結果相結合,也進一步證明SMA與MPEG和C12 H250H通過酯化反應制備出了目標產物。同樣通過6=6.41~7.32的質子峰面積與6=0.90、占=1.22—1.37和6=3.54的質子峰面積的比值,可以得到MPEG和C12H250H在SMA-g-MPEG+C12H250H中的摩爾分數為34.5%。
對于SMA-g-C12 H250H,譜圖中62.51(e)、3.43(c)、0.90(g)、6.41~7.32(a)和1.22~1.37(f、1)處化學位移的歸屬與SMA-g-MPEG+ C12 H250H譜圖中相同,同時還出現了63.63(h)處十二醇上與羥基相鄰的亞甲基中H的化學位移。1HNMR和FTIR的分析結果相結合,進一步證明SMA與C12H250H通過酯化反應制備出了目標產物。同樣通過6=6.41~7.32的質子峰面積與6 =0.90、6=1.22—1.37和6=3.63的質子峰面積的比值,可以得到Cl2H250H在SMA-g-C12H250H中的摩爾分數為36.7%。
凝膠滲透色譜法( GPC)測定SMA和接枝聚合物的分子量及其分布,結果如表1所示。
由表1可知,接枝聚合物的分子量大于SMA的分子量,進一步確定SMA和MPEG或十二醇發生了接枝反應。由GPC結果分析可知,通過“一鍋”法制備出了分子量適中,分散度較窄的梳型兩親聚合物。
2.3梳型兩親聚合物的性能
3種梳型兩親聚合物的水溶性對比見表2。
通過對比可知,樣品A、C、B的水溶性依次逐漸降低。樣品B、C在達到一定質量濃度后,便會形成凝膠,原因可能是十二醇的引入使樣品B、C的疏水性變強,大分子間存在極強的疏水相互作用、靜電力和范德華力,容易發生卷曲和相互纏繞,產生具有一定強度且可逆的物理締合,從而形成水分子包含在其中的三維立體空間網狀結構,宏觀上便表現成凝膠形式,其中樣品B的支鏈只是疏水性的十二醇,其疏水性更強,比樣品C更易形成凝膠,水溶性最差;而樣品A的支鏈只是親水性的MPEG,其上含有較多的乙氧基團,可以和水分子間形成很強的氫鍵,而大分子間的各秤相互作用則較弱,難以形成相互纏結的空間網狀結構,故其水溶性最好。
3種梳型兩親聚合物的表面張力曲線見圖3。
通過對比可知,樣品A降低表面張力的能力有限,其表面張力曲線上沒有出現平臺,不具備表面活性劑的基本特性。這一現象在其他兩親聚合物的研究中也有出現,說明此梳型兩親聚合物在表面的富集和在體相中的聚集行為均不同于小分子兩親物質,前者在CMC之前就開始有聚集體形成,而當溶液質量濃度達到CMC以上時,其在氣液界面的吸附仍在進行。此外,由于它帶有較長的乙氧基親水側鏈,側鏈間的相互作用使疏水主鏈在氣液表面形成穩定單層吸附和較為伸展的構象比較困難,這些都可能導致其表面張力曲線在高質量濃度區仍在變化。樣品B和C的表面張力曲線上出現平臺,且最后表面張力均達到較低值,這說明接枝十二醇后,梳型兩親聚合物的表面活性有較大提高。對其拐點進行擬合,可得樣品B的CMC為
3結論
(1)采用“一鍋”法制備了分子量適中,分散度較窄的梳型兩親聚合物SMA-g-MPEG、SMA-g-C12H25OH和SMA-g-MPEG+C12 H250H,并利用FTIR和1HNMR確定了其結構。
(2) SMA-g-MPEG的水溶性最好,在其質量濃度
(3) SMA-g-MPEG水溶液的表面活性較差,表面張力曲線沒有出現平臺,不具備表面活性劑的基本特性;SMA-g-MPEG+C12H250H水溶液的表面活性有所提高,表面張力曲線上出現平臺,其CMC為